

Sickle Cell Disease

Sickle Cell Disease (SCD) is caused by a haemoglobin defect, a structural variant known as Haemoglobin S, which replaces both β-globin subunits in haemoglobin.
This variant haemoglobin is an altered haemoglobin molecule, which when exposed to an environment low in oxygen, it sticks together to form long rods inside the red blood cells making these cells rigid and sickle-shaped; the process in medical terms is called polymerisation of the haemoglobin molecule.
Normal red blood cells are shaped like a donut and can bend and flex easily. The cells which become sickle shaped are rigid and have difficulty passing through small blood vessels, where they can get stuck and clog the blood flow (see Figure 1). This causes pain that can start suddenly, be mild to severe, and can last for any length of time. Because of this sickled shaped cells can block the blood supply to tissues, leaving them unable to oxygenate.
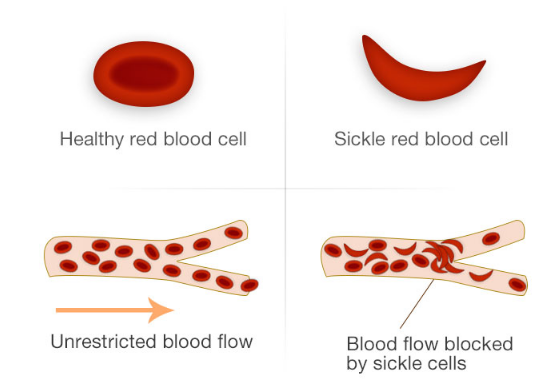
Figure 1
Such events can be severe enough to damage tissues in joints, spleen, kidneys, in fact all vital organs including the brain. In addition, these altered red cells (sickle cells) do not survive in the circulation for as long as normal cells do and are continuously destroyed. This causes patients to experience a degree of anaemia, which may become severe under certain circumstances, leading to a need for blood transfusion.
As a chronic disorder, sickle cell disease requires treatment in specialized centres aimed at both preventing and managing complications, including the prevention of infections through immunizations and the management of pain, which may be severe enough to require hospitalization. To prevent some of the complications effectively, it is necessary to have the patient under continuous observation from early childhood, and in this context a policy of newborn screening is recommended, so that affected children may be identified and followed up from birth.
The sickle cell condition can be caused when a person inherits the sickle cell gene from both parents (HbS/HbS) (see Figure 2) or if co-inherited with HbC or HbD or OArab (other variants) and with β- thalassaemia (see Figure 3).
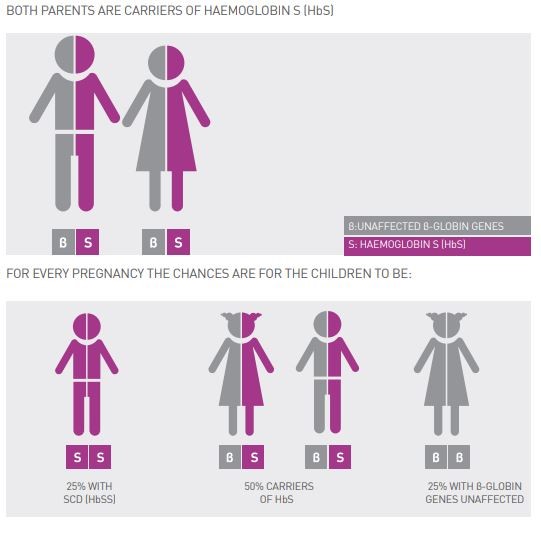
Figure 2

Figure 3
It is believed that the sickle cell abnormal haemoglobin originated in Africa, where it is most commonly encountered, while India is considered as an additional place of origin. HbS is prevalent also in the indigenous population of the Arab world and some Mediterranean countries (parts of Greece, Turkey, and Southern Italy).
In the past the slave trade had transported African populations to North and South America, so it is common in the USA, Brazil and the Caribbean islands. In more recent times, migrations have taken the gene to almost all regions of the world, especially Western and Northern Europe.
According to current epidemiological data, about 7% of the global population carries an abnormal haemoglobin gene, with more than 500,000 affected children born annually. More than 70% of them have a sickle disorder, and the rest have thalassaemia syndromes. A significant number of affected children born in developing countries even today die undiagnosed or misdiagnosed, receiving sub-optimal treatment, or left untreated altogether.
Newborn or neonatal screening:
Newborn or neonatal screening can identify both carriers of SCD, but also affected patients.
Children with sickle cell disease must be identified at birth through a special test and be offered special medical care early on to help prevent complications. With early diagnosis and treatment, together with parental education and involvement, it has been shown that early complications can be prevented and children have a better chance of survival. Sickle cell disease is becoming a chronic disease compatible with good survival. This is achieved by interventions like penicillin prophylaxis and vaccinations to prevent infections as well as careful follow up in specialised clinic.
To identify patients as soon as they are born, newborn screening programmes are being increasingly adopted across the world, including many countries where sickle cell disease has been introduced by recent migrations.
A whole variety of complications can occur in this condition and often these occur suddenly following a painful crisis. The signs and symptoms of sickle cell disease vary greatly from one person to another. Some affected people are quite healthy and are diagnosed at a relatively old age; others are frequently hospitalised and have many complications, while some die at an early age from the disease and its complications. The reasons for this marked variability in the clinical spectrum of this disease are not all known.
Main complications can include:
Painful crises are the commonest manifestation of sickle cell disease at all ages and dominate the clinical picture of sickle cell disease. These are usually acute and often very severe.
In Infancy or early childhood:
The first manifestation of sickle cell disease in infants is dactylitis or hand-foot syndrome which is a painful swelling and redness of hands and feet. In addition, infants and young children with sickle cell disease are extremely vulnerable to life-threatening infections in the lungs (pneumonia), blood (sepsis), lining of the brain (meningitis), and bone (osteomyelitis). Children under the age of five are at highest risk for these infections. The most worrisome infections are caused by a few types of bacteria, including Streptococcus pneumoniae (pneumococcus), Haemophilus influenzae type b (Hib), Neisseria meningitidis (meningococcus) and Salmonella. Other infections that children with sickle cell disease are vulnerable to are those caused by flu viruses.
Acute Splenic Sequestration is a leading cause of death in children with sickle cell disease and is a medical emergency. Most cases occur between 3 months to 5 years of age. In this condition, the spleen rapidly entraps blood leading to sudden onset of severe anaemia, circulatory collapse, and death in a few hours if not promptly detected and treated. Affected children present with acute onset of severe pallor, shock, and painful left-sided abdominal distension (bloating) with an enlarged and often massive spleen. Acute splenic sequestration has a high recurrence rate, particularly in infants below 1 year of age.
In most children, the spleen stays enlarged for the first few years of life but by 6 years of age, it usually becomes small and non-functioning due to scarring from recurrent sickling and multiple infarctions. That is why acute splenic sequestration is usually infrequent after 6 years of life.
In older children, adolescents, and young adults:
Acute Chest Syndrome is the first cause of early death and the second cause of hospitalization in patients with sickle cell disease. It is caused by trapped sickle cells in the blood vessels of the lungs or by an infection or a fat or bone marrow embolus. About 50% of cases of acute chest syndrome occur a few days after hospitalization with acute painful crises.
Overt stroke is seen mostly in young children with sickle cell disease and mostly in those with sickle cell anaemia. The best way to know if a child is at high risk for getting a stroke is by a special test called Transcranial Doppler (TCD). This test measures the velocity of blood flow to the brain. When a high velocity of blood is indicated in the brain vessels, a child is at high risk of getting a stroke. Often stroke can be silent, that is without clinical manifestations.
Less frequently and more so in adults, stroke can be due to bleeding in the brain (haemorrhagic stroke).
A serious outcome of anaemia, strokes, and silent brain infarcts is the development of neurocognitive problems. This is often an underdiagnosed complication and is detected by special tests that assess intelligence, memory, and comprehension (neuropsychiatric and neurobehavioral testing) and not by imaging of the brain or blood vessels of the brain. To identify this complication early, all children with sickle cell disease should be screened with routine exams, starting at 6 years of age.
Acute Anaemia: The majority of patients with sickle cell disease (SCD) have some degree of baseline anaemia due to premature death (haemolysis) of the sickle red blood cells. Symptoms of anaemia include pallor (yellow skin colour), getting tired easily, irritability, headache, loss of appetite, and poor growth. People with sickle cell disease can also get acute (sudden onset) anaemia due to:
- Blood becoming suddenly entrapped in the spleen (acute splenic sequestration)
- Sudden cessation of blood cell production (aplastic episode) due to certain infections. This means a sudden cessation of blood cell production. It can be caused by infections most commonly parvovirus B19.
- Excessive red blood cells breakdown (hyper-haemolytic crisis). The increased haemolysis can occur during an episode of pain, infection, drug exposure or can be due to an acute or delayed reaction to a red cell transfusion.
Avascular Necrosis usually occurs between the ages of 15 and 50 years and is not seen frequently in young children. This condition is seen when the blood flow to body areas with poor baseline blood supply is slowed and obstructed by sickle cells, leading to tissue breakdown (necrosis). It occurs mostly in the hip (femoral head) which suffers a loss of blood flow (avascularity) due to obstructive sickle cells. Another vulnerable joint is the shoulder (humeral head).
Osteomyelitis, an infection of the bones, affects mostly the long bones of the legs, thighs and arms and is often a complication of leg ulcers. The 2 most frequent bacteria seen in osteomyelitis are Salmonella, which causes typhoid fever and gastro-enteritis, and Staphylococcus aureus.
Osteoporosis or weak bones with low bone mineral density (BMD) is a very frequent complication seen in 30 to 80% of patients with sickle cell disease. It is often asymptomatic and affects mostly the spine. Osteoporosis may lead to fractures (seen in 15-30% of patients with SCD) of long bones and bony pain and deformities.
Leg Ulcers are painful lesions around the ankle, seen in 10 to 20% of sickle cell anaemia patients, and usually appear between 10 and 50 years of age.
Sickle cell disease affects the gall bladder, the liver, and the small and large tubes that carry bile inside and outside the liver (bile ducts). These are mostly due to recurrent episodes of blood flow blockage by sickle cells and increased bilirubin due to red cell haemolysis leading to gallstones. Other causes of liver disease in sickle cell disease include viral infections and increased iron in the liver due to infected blood transfusions.
Kidney problems in sickle cell disease often start in childhood and rarely in infancy. The usual tests of kidney function are often normal, even in the face of existing chronic kidney disease, until extensive kidney damage has occurred. The kidney can be affected in sickle cell disease in various ways: Obstruction of blood flow by sickled RBCs; inability to concentrate their urine so that patients pass more urine than normal and continue to have bedwetting to older age; infections of the bladder and kidney are quite common, particularly during pregnancy. With older age and repetitive clogging of the blood vessels that nourish the kidneys, the glomerulus (the part of the kidney that filters waste products from the blood and initiates urine formation) may be damaged and renal failure can develop. This is usually preceded by excessive loss of a protein (albumin) in the urine.
Priapism is a sustained unwanted painful erection of the penis seen in around 35% of boys and men. Priapism is seen at any age and is due to decreased blood flow and oxygen in the penis due to sickling. Over time, priapism can damage the penis and lead to partial or total impotence.
Eye Problems: blockage of blood flow by sickle cells can affect any part of the eye and lead to several complications including bleeding, scarring, and rarely blindness. The back of the eye (retina), which is the most important part of vision, is most sensitive to this blockage because it contains tiny blood vessels.
With age, more patients with sickle cell disease will need transfusions to treat and prevent complications other than stroke. Blood transfusions will inevitably lead to iron overload. People who undergo regular transfusions need to be closely monitored for iron overload and must receive early iron removal treatment (chelation) to reduce iron levels.
Treatment is usually aimed at avoiding crises, relieving symptoms and preventing complications. Babies and children age 2 and younger with sickle cell anaemia should make frequent visits to a doctor. Children older than 2 and adults with sickle cell anaemia should see a doctor at least once a year. Treatments might include medications to reduce pain and prevent complications, and blood transfusions, as well as a bone marrow transplant.
Vaccinations:
Childhood vaccinations are important for preventing disease in all children. They’re even more important for children with sickle cell disease because their infections can be severe. Particularly important is the immunization of children with the 7-valent pneumococcal conjugate vaccine in addition to the 23-valent polysaccharide pneumococcal vaccine. Also meningococcal vaccination and Haemophilus influenzae type b (Hib), according to the national vaccination schedule in each country. Hepatitis B vaccine should not be forgotten by potential recipients of blood transfusion. Annual influenza vaccination after six months of age is also recommended (Mehta SR et al. Am Fam Physician. 2006 Jul 15;74(2):303-310).
Antibiotics for the Prevention of Infections:
Children with sickle cell disease should begin taking the antibiotic penicillin when they’re about 2 months old and continue taking it until they’re at least 5 years old. Doing so helps prevent infections, such as pneumonia, which can be life-threatening to an infant or child with sickle cell anaemia. As an adult, if the spleen was removed or had pneumonia (acute chest syndrome), penicillin should be taken throughout life.
Hydroxyurea:
When taken daily, hydroxyurea reduces the frequency of painful crises and might reduce the need for blood transfusions and hospitalizations. Hydroxyurea seems to work by stimulating the production of fetal haemoglobin — a type of haemoglobin found in newborns that helps prevent the formation of sickle cells.
Blood Transfusions:
Donated blood will increase the number of normal red blood cells in circulation, helping to relieve anaemia, but helps also to reduce red cell production by the patient’s own blood-forming tissue in the bone marrow and this will also reduce the production of sickle cells. In children with sickle cell anaemia at high risk of stroke, regular blood transfusions can decrease the risk. Transfusions can also be used to treat other complications of sickle cell disease, or they can be given to prevent complications. Many patients are now on regular transfusions for these reasons but of course, they have to face the possible complications of transfusions, including accumulation of iron, allo-immunisation, and viral hepatitis. Monitoring of iron overload, as in thalassaemia, with a possible need for iron chelating medication becomes an important part of management.
Bone Marrow Transplantation (BMT), also called Haemopoietic Stem Cell TransplantATION (HSCT):
This involves replacing bone marrow affected by sickle cell anaemia with healthy bone marrow from a donor. The procedure usually uses a matched donor, such as a sibling, who doesn’t have sickle cell anaemia. For many, donors aren’t available (see the BMT section on thalassaemia). This procedure is recommended for children or young patients with severe disease.
Voxelotor (Oxbryta™):
Voxelotor (Oxbryta), an HbS polymerization inhibitor, was granted approval by the U.S. Food & Drug Administration (FDA) in 2019 for the treatment of SCD in adults and pediatric patients 12 years and older. In February 2022, the EC also granted marketing authorization for Oxbryta for the treatment of haemolytic anemia due to SCD. Since its first authorization in 2019, the drug has been approved in over 35 countries globally.
In September 2024, the EMA’s Human Medicines Committee recommended suspending the marketing authorization of Oxbryta, citing safety and efficacy concerns that emerged during a review of Oxbryta after data from a clinical trial showed that a higher number of deaths occurred with the drug than with placebo and another trial showed the total number of deaths was higher than anticipated.
Crizanlizumab (Adakveo®):
Crizanlizumab (Adakveo) was granted approval by the U.S. Food and Drug Administration (FDA) in November 2019 to reduce the frequency of VOCs (pain crises) in individuals with sickle cell disease. The therapy had been conditionally approved in Europe in November 2020 and in the U.K. in October 2021 for similar indications. It is a monoclonal antibody designed to block P-selectin, a protein found on blood vessel cells that contributes to the clumping of sickled red blood cells and their adhesion to blood vessel walls. In blocking that protein, the treatment was expected to improve blood flow and reduce the frequency of VOCs.
A Phase 3 clinical trial called STAND (NCT03814746) is testing the safety and efficacy of Adakveo at the now-approved 5 mg/kg dose and at a higher dose of 7.5 mg/kg, both against a placebo, in more than 250 adults and adolescents, ages 12 and older, with SCD. After one year of treatment, neither dose outperformed a placebo at reducing the annual rate of VOCs requiring a healthcare visit. While no new safety concerns were identified, patients on Adakveo experienced more serious side effects than those on a placebo.
These findings, inconsistent with the results that supported Adakveo’s approval in the U.S., led the European Commission in August 2023 to revoke the therapy’s conditional marketing approval. The U.K. Medicines & Healthcare products Regulatory Agency (MHRA) also withdrew its authorization to market the medication. In the US, the therapy remains approved for the treatment of VOCs in adults and pediatric patients, ages 16 and older.
The STAND trial is expected to finish in 2026.
Gene editing therapies:
In December 2023, the FDA approved two gene-editing treatments for patients aged 12 and older. The first therapy, Exagamglogene autotemcel (Casgevy), developed by Vertex Pharmaceuticals and CRISPR Therapeutics, utilizes the innovative CRISPR gene-editing tool. With Casgevy, an edit (or “cut”) is made in a particular gene to reactivate the production of fetal haemoglobin, which dilutes the faulty red blood cells caused by sickle cell disease. The second treatment, Lovotibeglogene autotemcel (Lyfgenia) by Bluebird Bio, employs a different gene-editing technique using a lentiviral vector to deliver a healthy haemoglobin-producing gene.
The therapies are hailed as groundbreaking as they represent the first-ever gene therapies to potentially cure a hereditary condition. Nevertheless, widespread availability is not anticipated initially. The cost of these cutting-edge treatments is estimated to be between $2 million and $3 million per patient, which may limit accessibility at the outset, and are available only at large, authorized medical centers because they require advanced care.
To date, Casgevy has received marketing authorisations by regulatory authorities in the EU, the UK, Saudi Arabia, Bahrein, and Canada. Lyfgenia has been given orphan drug and priority medicines designations in the EU.
Patients who received Casgevy or Lyfgenia will be followed in a long-term study to evaluate each product’s safety and effectiveness.
Also see:
Charter for Optimal Care Transitions from Paediatric to Adult Care in Sickle Cell Disease
Guideline for the Emergency Department for the Acute Treatment of Patients with Sickle Cell Disease